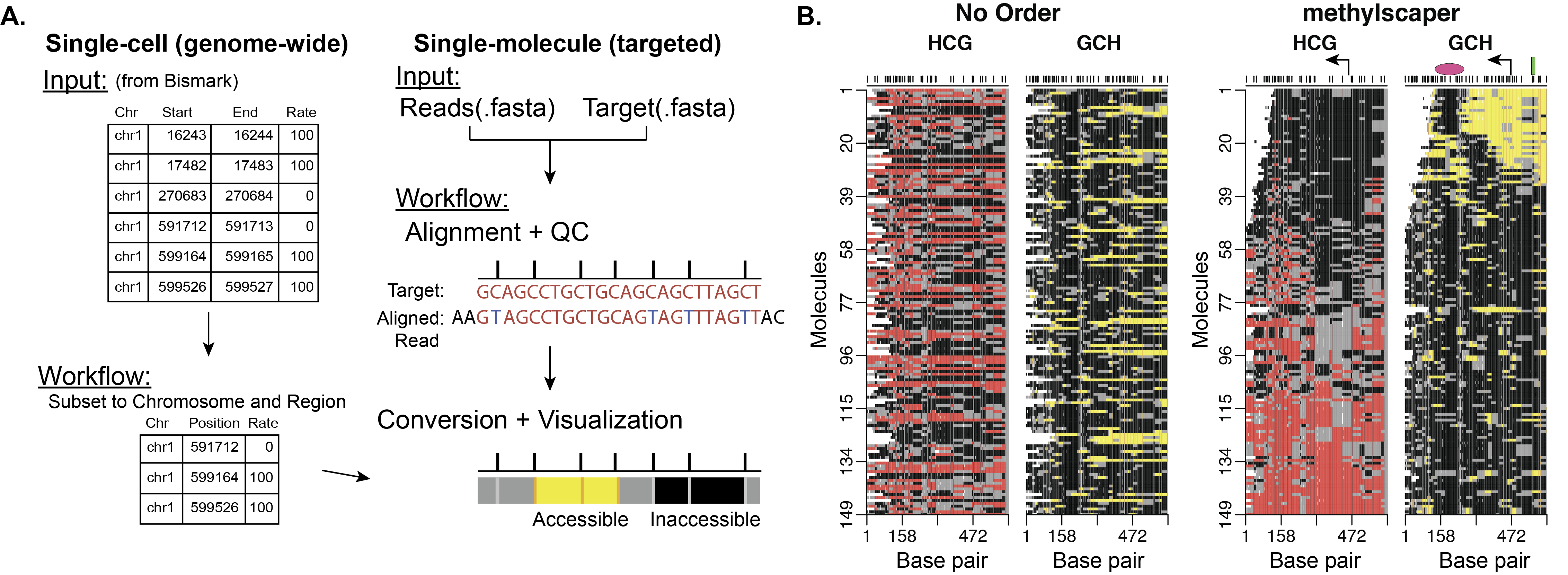
methylscaper is an R package for visualizing data that jointly profile endogenous methylation and chromatin accessibility (MAPit, NOMe-seq, scNMT-seq, nanoNOMe, etc.). The package offers pre-processing for single-molecule data and accepts input from Bismark (or similar alignment programs) for single-cell data. A common interface for visualizing both data types is constructed by generating ordered representational methylation-state matrices. The package provides a Shiny app to allow for interactive and optimal ordering of the individual DNA molecules to discover methylation patterns, nucleosome positioning, and transcription factor binding.
| Manuscript | Vignette | Example Data | FAQ | Contact |
The Shiny app may be run locally following the instructions in the vignette, or we have a web-based version of the app available:
Start Shiny App